
Le problème du repliement protéique, c’est à dire de la détermination de la forme tridimensionnelle détaillée d’une protéine, a occupé une place centrale dans la science des protéines tout au long du 20ème siècle. En effet, ces informations structurales sont essentielles pour nous permettre de comprendre comment une protéine fonctionne, et donc comment améliorer son activité, ou au contraire l’entraver (si cette protéine est impliquée dans une pathologie).
Après la détermination par cristallographie des structures de l’hémoglobine et de la myoglobine en 1960, les chercheur·se·s du monde entier ont rivalisé d’ingéniosité pour obtenir la structure de systèmes de plus en plus gros et complexes, via le développement de nouvelles méthodes expérimentales, telles que la RMN ou la microscopie cryo-électronique. Ces précieuses informations sont stockées sur la Protein Data Bank, un base de données qui fête ses 50 ans en 2021 et qui héberge actuellement plus de 170 000 structures expérimentales de proteines (et aussi quelques milliers de structures d’ADN et d’ARN parce qu’il ne faut pas être sectaire).
Cet atlas du vivant à l’échelle moléculaire a beau être gigantesque, il existe encore des milliers de protéines dont on ne connait que la séquence (soit l’enchainement des acides aminés), mais dont la structure résiste encore et toujours aux expérimentateurs·rices. Et c’est là que les groupes de recherche en modélisation entrent en scène. Créée en 1994, la compétition CASP (Critical Assesment of protein Structure Prediction) propose aux équipes de théoricien·ne·s de prédire la structure de protéines pour lesquelles les données expérimentales n’ont pas encore été dévoilées publiquement. À chaque round de la compétition (qui se tient tous les deux ans) les modèles proposés par les équipes participantes sont comparés à la structure expérimentale de référence et se voient attribuer une note comprise entre 0 (copie à revoir intégralement) et 100 (prédiction parfaitement conforme à l’expérience). Chaque round propose de déterminer les structures de plusieurs protéines (les cibles) avec des niveaux de difficulté variables. Ainsi, lors de la première édition de CASP, les notes moyennes des modèles proposés pour les différentes cibles s’étalaient de 80% (petite protéine facile à modéliser) à 20% pour les systèmes les plus complexes, avec une note moyenne globale autour de 40%. Au fil des ans et des améliorations dans les programmes de modélisation, cette performance globale s’est bien sur améliorée jusqu’à atteindre une note moyenne de 75% en 2020.
La présentation des résultats de l’édition 2020 de CASP le 30 novembre dernier a eu un écho tout particulier auprès de la communauté scientifique (et même du grand public), car l’une des équipes participante a proposé des prédictions de structures très nettement supérieures à celles de ses compétitrices. Le logiciel AlphaFold2 (qui est lui même une amélioration de AlphaFold1, qui avait déjà remarquablement bien fonctionné lors de CASP13 en 2018) développé dans le laboratoire DeepMind (qui appartient à Google) obtient en effet une note moyenne globale autour de 90%. Ce qui signifie que les modèles produits par cette équipe sont comparables à ceux obtenus par des méthodes expérimentales. D’ailleurs dans certains cas les groupes expérimentaux ont même corrigé leur modèle sur la base des prédictions faites par AlphaFold2. Ce logiciel s’appuie sur une approche de type Deep Learning (apprentissage profond)/Intelligence artificielle qui repose sur une base de données pour apprendre comment les protéines se replient. Ici la base de données c’est bien sur la PDB et ses 170 000 structures, qui vont fournir une énorme quantité d’informations, par exemple quels acides aminés sont susceptible de se rapprocher lorsque la protéine se replie. AlphaFold2 utilise également des données évolutives, en regroupant les protéines avec des séquences proches, et qui ont alors de fortes chances de présenter des structures similaires. Dans l’ensemble, les extraordinaires résultats produits par le logiciel ont amené beaucoup de scientifiques (et de journalistes à leur suite) à déclarer que le problème du repliement protéique est désormais résolu.
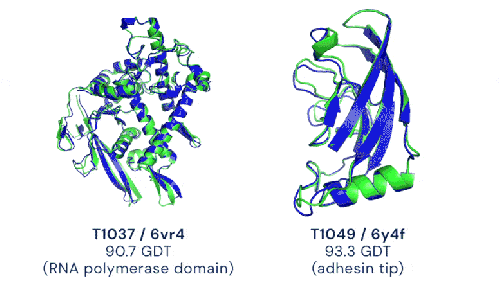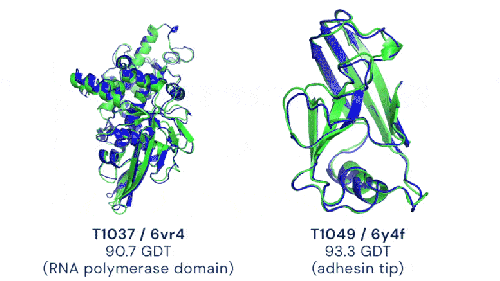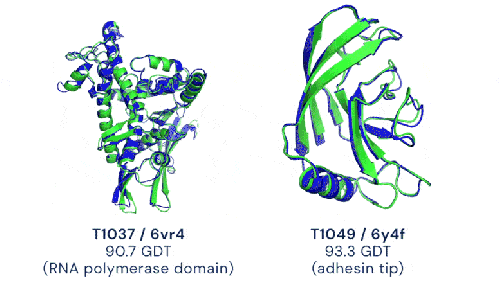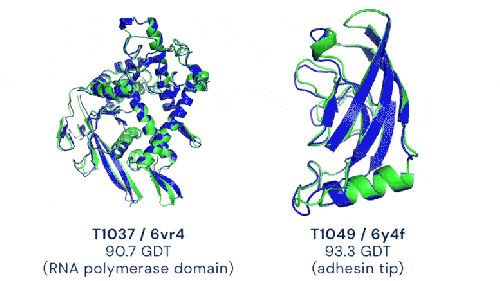
Deux exemples de prédictions d’AlphaFold2 (en bleu) comparées à la structure expérimentale de référence (en vert) avec des notes supérieures à 90% (image de DeepMind)
Et maintenant ? On remballe tout et on rentre à la maison ?
Pas si vite papillon !
Une caractéristique des méthodes de type Deep Learning, c’est qu’elles sont limitées par la base d’apprentissage qu’on leur fournit. En gros l’algorithme va assimiler toutes les informations présentes dans la base de données (ce qui est déjà remarquable), mais n’en produit pas de nouvelles. En l’occurence, la collection de 170 000 structures présentes dans la PDB présente trois limitations d’importance sur le plan biologique :
- Il s’agit principalement de protéines isolées. Or dans la cellule les protéines fonctionnent majoritairement en interaction les unes avec les autres. Elles vont former de grands assemblages, des complexes, où chacun des partenaires est susceptible de se déformer localement au niveau du point d’assemblage. Les structures des complexes protéiques sont encore minoritaires (« seulement » 30 000 structures disponibles) et AlphaFold2 a donc bénéficié de moins de données d’apprentissage pour ce type de système, ce qui peut lui poser problème. Heureusement pour les biologistes structuraux, la compétition CASP a une petite cousine, CAPRI (Critical Assessment of PRediction of Interactions), qui s’intéresse justement à la prédiction des interactions protéiques.
- Il s’agit principalement de protéines repliées. Par définition, pour qu’une protéine figure dans la PDB, cela signifie que que l’on a pu résoudre sa structure expérimentalement. Mais il existe également des protéines qui ne se replient pas (soit dans leur intégralité, soit au niveau des certains fragments). Ces IDP (Intrisically Disordered Proteins/Protéines Intrinsèquement Désordonnées) ont beau être sous-représentées dans la PDB (on a longtemps parlé de matière noire du monde protéique à leur sujet), elles ne représentent pas moins de 30% du protéome des eucaryotes. Et pour comprendre leur fonctionnement, il est impossible de se limiter à une seule structure, aussi précise soit-elle, mais il faut plutôt s’intéresser à leur propriétés dynamiques, soit la façon dont elles peuvent changer de forme au cours du temps, ce qu’AlphaFold2 est encore bien incapable de faire.
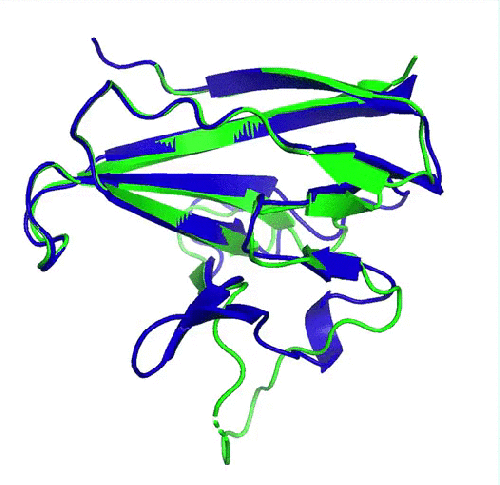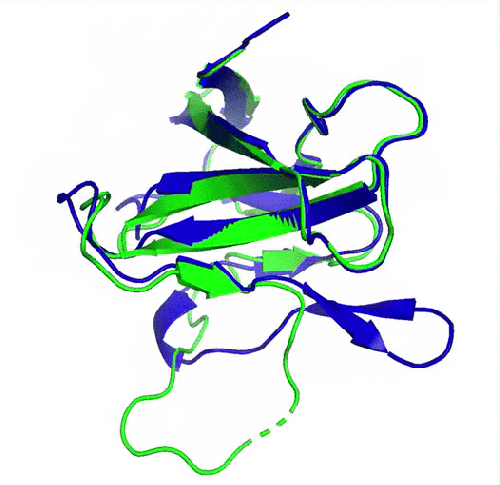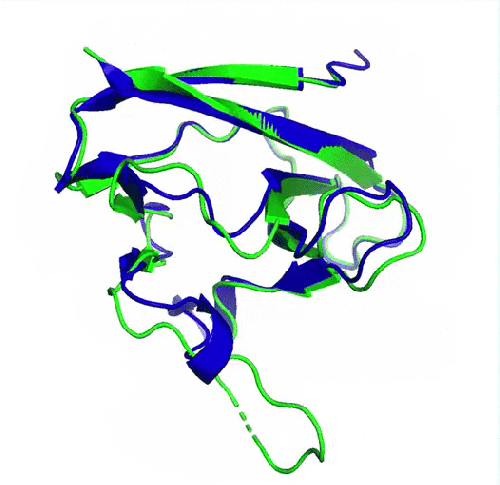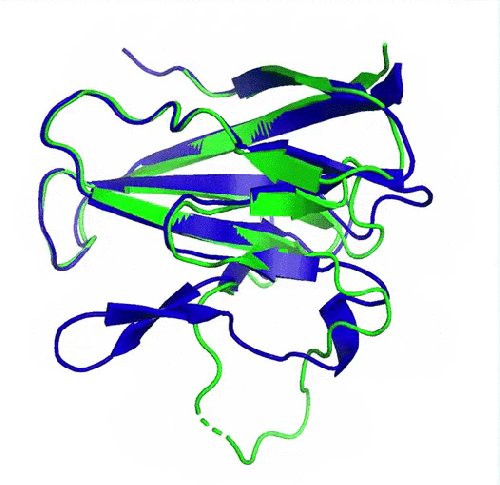
Prédiction de la protéine ORF8 du SARS-CoV2 (en bleu) comparée à la structure expérimentale de référence (en vert, pdb 7jtl)). Le plus grand écart entre prédiction et expérience se situe au niveau d’une boucle flexible (en bas de la protéine) et donc dépourvue de structure secondaire. (image de DeepMind)
- Il manque encore les modifications post-traductionnelles. Acétylation, glutamylation, phosphorylation… ces modifications chimiques des acides aminés qui ont lieu après la fabrication de la protéine dans le ribosome sont susceptibles de modifier la structure et la fonction protéique. Impossible donc de s’arrêter au seul repliement pour pour tout savoir de l’activité d’une protéine !
Au final AlphaFold2 n’en représente pas moins une avancée extraordinaire dans notre connaissance des protéines, mais cela reste une étape (certes importante) sur la longue route à parcourir pour comprendre le vivant à l’échelle moléculaire.
Pour en savoir plus :
- Un billet avec plus de détails techniques (notamment sur l’algorithme développé dans AlphaFold) sur le blog de David Louapre.
- Une note de blog très détaillée (en anglais) d’un groupe de recherche d’Oxford qui revient sur les résultats d’AlphaFold.
- Le communiqué de presse (en anglais) de DeepMind sur AlphaFold2
- Une bande dessinée de SMBC sur les véritables motivations de DeepMind.
- Un épisode de la Science CQFD (mars 2024) sur AlphaFold2
- En octobre 2024, Demis Hassabis et John Jumper, deux des concepteurs d’AlphaFold2, ont reçu le Prix Nobel de Chimie.
