
Les protéines présentent une très grande variété de formes et de tailles. Celles-ci peuvent en effet être constituées de 50 à plus de 30 000 acides aminés (pour l’immense titine que l’on peut trouver dans les muscles), et si on s’intéresse aux très grands assemblages de plusieurs protéines on peut même monter à plus de 300 000 acides aminés dans les enveloppes de virus. En biochimie, on décrira la structure d’une chaîne protéique en distinguant quatre niveaux d’organisation :
- Tout commence en suivant le plan fourni par l’ADN. Le ribosome (qui est lui même un énorme assemblage de protéines avec laquelle vous pourrez faire plus ample connaissance prochainement) va lire la séquence de nucléotides de l’ADN et la déchiffrer grâce au code génétique. Cela donne l’enchaînement des acides aminés, ou résidus, constitutifs de la protéine, que l’on va appeler la structure primaire, ou encore la séquence protéique. Les séquences protéiques ont un sens de lecture (qui indique l’ordre dans lequel les résidus ont été associés les uns aux autres par le ribosome). On démarre toujours par un atome d’azote (au niveau de l’extrémité que l’on appelle donc N-terminale, N étant le symbole de l’azote), qui correspond à la fonction amine (NH2) portée par le premier acide aminé, et on conclut par un atome de carbone (au niveau de l’extrémité C-terminale) de la fonction acide (COOH) du dernier acide aminé de la chaîne protéique.
- La structure secondaire décrit les arrangements locaux de ces acides aminés qui sont généralement stabilisés par des liaisons hydrogène (cf. ce billet). Boucles, hélices α ou feuillets β , plusieurs éléments de structure secondaire distincts peuvent coexister au sein d’une même chaîne protéique.
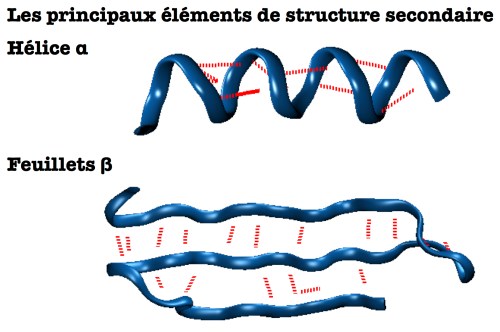
- La structure tertiaire décrit comment plusieurs éléments de structure secondaire vont s’agencer les uns par rapports aux autres au sein d’un domaine protéique. Pour décrire la forme d’ensemble d’une protéine, qui peut donc comporter plusieurs domaines et fluctuer au cours du temps, on parlera de conformation ou de repliement protéique.
- Finalement la structure quaternaire définit l’assemblage au sein d’un complexe de plusieurs chaînes protéiques déjà repliées (que l’on appellera alors des sous-unités). Toutes les protéines ne comportent pas forcément plusieurs sous-unités et donc de structure quaternaire.
C’est quoi une protéine native ?
On parle de protéine native, ou encore de forme sauvage de la protéine, lorsque sa séquence est celle produite majoritairement par une population d’organismes vivants, et de variant (ou mutant), lorsque cette séquence native a été modifiée ponctuellement au niveau de l’ADN, et donc de la protéine.
Une mutation peut être :
- Spontanée La mutation d’un acide aminé vers un autre dans la séquence d’une protéine peut se produire spontanément : c’est le moteur de l’évolution avec la sélection naturelle. En effet la sélection naturelle se fait sur une population dans laquelle il y a des différences et favorise les individus qui sont les plus adaptés à un environnement et à un moment donnés.
- Artificielle Les mutations peuvent également être provoquées lors d’expériences en laboratoire, il s’agit alors d’évolution dirigée.
Les modifications de la séquence d’une protéine peuvent être sans conséquence sur celle-ci, dans ce cas on parlera de variant d’une protéine quand il n’y a pas de pathologie associée à une mutation. Mais parfois, au contraire, les variations de séquence peuvent avoir un fort impact sur la structure et l’activité protéiques, et pour en savoir plus là dessus, il faut faire un tour au chapitre \ref{mutant}.
Enfin, on appelle structure native la structure d’une protéine qui lui permet de réaliser une fonction biologique donnée (cf. le billet suivant). Lorsque cette structure est modifiée (par exemple sous l’effet de la chaleur ou suite à une réaction chimique), on parlera alors de dénaturation de la protéine.
Vous pensez pouvoir déterminer la structure native d’une protéine ?
La prédiction d’une structure protéique (secondaire, tertiaire et éventuellement quaternaire si la molécule présente plusieurs chaînes) à partir de sa seule séquence d’acides aminés est un problème incroyablement complexe qui occupe les chercheur·se·s en biologie structurale depuis plusieurs décennies.
Les méthodes actuelles utilisent les outils développés en chimie théorique et en bioinformatique et sont régulièrement confrontées lors de l’expérience CASP (Critical Assesment of protein Structure Prediction) qui a démarré en 1994 et a lieu tous les deux ans. À cette occasion, des chercheur·se·s du monde entier travaillant sur le sujet entrent en compétition et doivent proposer les meilleurs prédictions de structure possibles pour des protéines.

Vous voulez tenter votre chance ?
C’est possible grâce à un projet de science citoyenne: le logiciel Foldit vous apprend à replier les protéines dans le cadre d’un jeu vidéo !
Les meilleur·e·s joueur·se·s de Foldit ont ainsi pu contribuer à la détermination de la structure d’une protéine virale que les chercheur·se·s et leurs ordinateurs n’avaient pas réussi à résoudre.
À l’origine, les chercheur·se·s sont partis du principe qu’une protéine devait forcément adopter une structure secondaire, et/ou tertiaire, bien définie pour être fonctionnelle. On sait désormais qu’il existe aussi toute une catégorie de protéines dites intrinsèquement désordonnées qui ne possèdent pas de structure native, ou qui ne vont se replier que dans le cadre d’une interaction avec d’autres biomolécules (protéine, ADN, membrane lipidique). Ces IDP (Intrinsically Disordered Proteins) sont pourtant parfaitement fonctionnelles et mêmes indispensables à de nombreux processus cellulaires. Bref, les chercheur·se·s ont encore du pain sur la planche avant d’épuiser le sujet du (non)-repliement protéique !

Awesome blog youu have here
J’aimeJ’aime